2025年4月15日,国际学术期刊Cell Death & Disease在线发表了高致病性病毒与生物安全全国重点实验室/武汉大学生命科学学院朱应、刘实团队的最新研究成果,论文题为“Hepatitis B surface antigen hijacks TANK-binding kinase 1 to suppress type I interferon and induce early autophagy”。 该研究工作揭示了乙型肝炎病毒表面抗原(HBsAg)通过劫持 TANK 结合激酶1(TBK1)抑制 I 型干扰素的产生并促进早期阶段自噬小体的积累, 为进一步理解乙型肝炎病毒(HBV)感染过程中宿主天然免疫应答和细胞自噬之间的相互关系提供了新的思路

HBV是一种嗜肝性的DNA病毒,可引起急性感染或慢性感染,进而发展成为不同程度的肝炎、肝硬化甚至肝癌。在前期的研究中,朱应、刘实团队发现,HBV感染机体后利用多种策略逃避宿主天然免疫系统识别;另一方面,HBV 的感染激活自噬并利用自噬促进病毒复制。在本研究中,该团队进一步探索了HBV调控宿主天然免疫应答和细胞自噬之间的分子机制。
在这项研究中,通过细胞和转基因小鼠动物模型研究团队发现HBsAg作为HBV的一种免疫逃逸蛋白,劫持干扰素信号通路关键分子TBK1抑制I型干扰素产生并促进自噬小体形成。进一步的机制研究显示,HBsAg在体外和体内模型与TBK1的激酶结构域(KD)相互作用增强其二聚化和磷酸化,同时破坏TBK1-IRF3复合物的形成,导致干扰素调节因子3 (IRF3)磷酸化受阻从而抑制I型干扰素产生;另一方面,活化的TBK1对HBV复制非常重要,这可能由于HBsAg增强TBK1二聚化通过TBK1-p62轴启动早期自噬促进了HBV的复制。此外,HBsAg还通过抑制突触体相关蛋白29(SNAP29)启动子活性从而阻断了自噬小体-溶酶体融合。在体内实验中,研究团队还发现HBs转基因小鼠或慢性HBV患者的肝组织内IFN-β信号传递被抑制,自噬小体形成被限制在早期。综上,本研究揭示了HBV调控宿主天然免疫应答和细胞自噬的新机制,这可能导致病毒持续感染。
武汉大学生命科学学院骆传进博士、马采娇博士为该论文的共同第一作者,朱应教授和刘实教授为本文共同通讯作者。本工作得到了国家重点研发计划(2021YFC2701800,2021YFC2701804)、国家自然科学基金(U22A20335)、湖北省杰出青年科学基金(2021CFA054)等项目的资助。
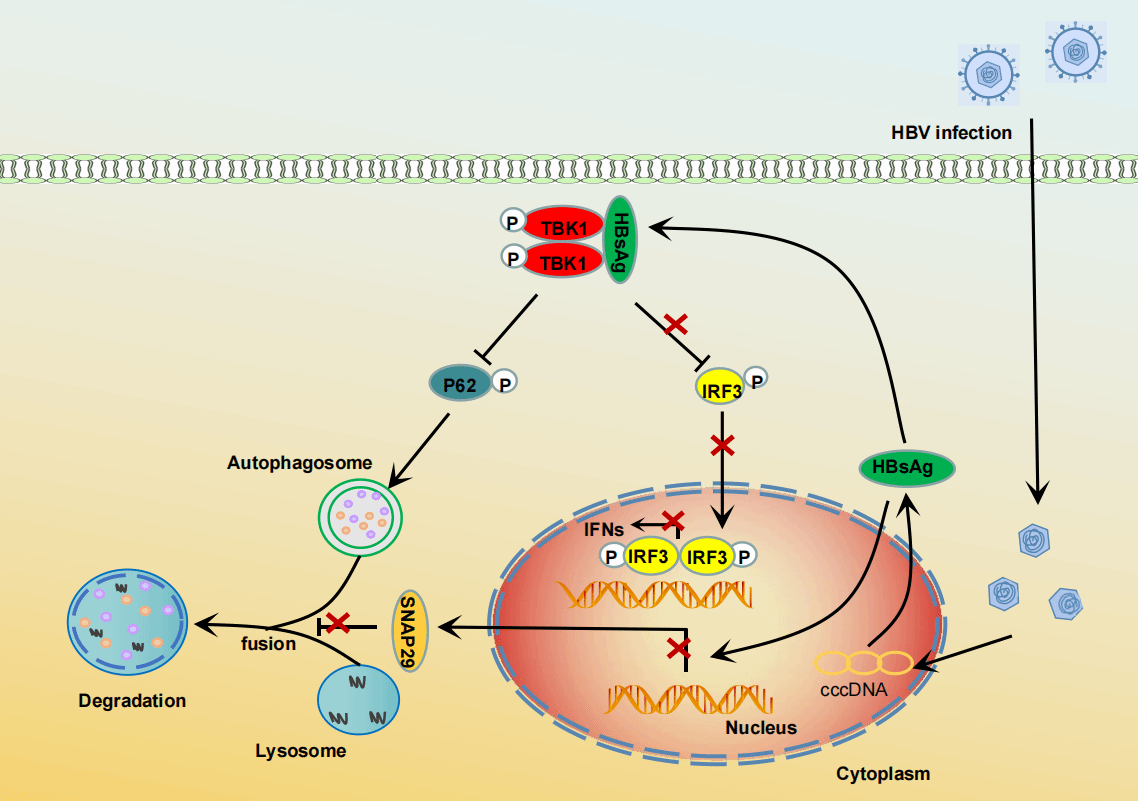
HBsAg宿主调控天然免疫应答和自噬模式图
论文链接: https://www.nature.com/articles/s41419-025-07605-0
